library(MicrobiomeDB, quietly = TRUE)
#> Warning: replacing previous import 'S4Arrays::makeNindexFromArrayViewport' by
#> 'DelayedArray::makeNindexFromArrayViewport' when loading 'SummarizedExperiment'
library(tidyverse, quietly = TRUE)
#> ── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
#> ✔ dplyr 1.1.4 ✔ readr 2.1.5
#> ✔ forcats 1.0.0 ✔ stringr 1.5.1
#> ✔ ggplot2 3.5.1 ✔ tibble 3.2.1
#> ✔ lubridate 1.9.3 ✔ tidyr 1.3.1
#> ✔ purrr 1.0.2
#> ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
#> ✖ dplyr::filter() masks stats::filter()
#> ✖ dplyr::lag() masks stats::lag()
#> ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errorsWhat is Alpha Diversity?
Alpha diversity measures the diversity of microbial taxa within a single sample or community. It takes into account both the number of different taxa (richness) and their distribution (evenness). Understanding alpha diversity provides insights into the complexity and structure of microbial communities at a local level.
Why Care About Alpha Diversity?
Researchers are interested in alpha diversity for several reasons:
Community Comparisons: Compare the diversity of microbial communities across different samples or conditions.
Health Assessments: Assess the health or stability of microbial communities within specific environments or host systems.
Ecological Understanding: Gain insights into the ecological dynamics of microbial communities at a local scale.
How is Alpha Diversity Calculated?
This package offers three diversity indices for calculation: Shannon, Simpson and Evenness.
Shannon Diversity Index
The Shannon diversity index measures the entropy or uncertainty in predicting the identity of a randomly chosen taxon within a sample.
It can be calculated as follows:
## first lets find some interesting data
microbiomeData::getCuratedDatasetNames()
#> [1] "Anopheles_albimanus" "BONUS"
#> [3] "Bangladesh" "DailyBaby"
#> [5] "DiabImmune" "ECAM"
#> [7] "EcoCF" "FARMM"
#> [9] "GEMS1" "HMP_MGX"
#> [11] "HMP_V1V3" "HMP_V3V5"
#> [13] "Leishmaniasis" "MALED_2yr"
#> [15] "MALED_diarrhea" "MORDOR"
#> [17] "Malaysia_helminth" "NICU_NEC"
#> [19] "PIH_Uganda" "PretermInfantResistome1"
#> [21] "PretermInfantResistome2" "UgandaMaternal"
getCollectionNames(microbiomeData::HMP_MGX)
#> [1] "Shotgun metagenomics 4th level EC metagenome abundance data"
#> [2] "Shotgun metagenomics Metagenome enzyme pathway abundance data"
#> [3] "Shotgun metagenomics Metagenome enzyme pathway coverage data"
#> [4] "Shotgun metagenomics Genus (Relative taxonomic abundance analysis)"
#> [5] "Shotgun metagenomics Species (Relative taxonomic abundance analysis)"
#> [6] "Shotgun metagenomics Family (Relative taxonomic abundance analysis)"
#> [7] "Shotgun metagenomics Order (Relative taxonomic abundance analysis)"
#> [8] "Shotgun metagenomics Phylum (Relative taxonomic abundance analysis)"
#> [9] "Shotgun metagenomics Class (Relative taxonomic abundance analysis)"
#> [10] "Shotgun metagenomics Normalized number of taxon-specific sequence matches"
#> [11] "Shotgun metagenomics Kingdom (Relative taxonomic abundance analysis)"
## grab a collection we like
genus <- getCollection(microbiomeData::HMP_MGX, 'Shotgun metagenomics Genus (Relative taxonomic abundance analysis)')
## get an alpha diversity ComputeResult
alphaDivOutput <- alphaDiv(genus, method = 'shannon')
#>
#> 2024-06-26 14:47:56.150053 Received df table with 741 samples and 226 taxa.
#>
#> 2024-06-26 14:47:56.202425 shannon alpha diversity computation complete.
#>
#> 2024-06-26 14:47:56.220225 Alpha diversity computation completed with parameters method= shannonSimpson Diversity Index
The Simpson diversity index measures the probability that two individuals randomly selected from the sample will belong to different taxa.
It can be calculated as follows:
## get an alpha diversity ComputeResult
genus <- getCollection(microbiomeData::HMP_MGX, 'Shotgun metagenomics Genus (Relative taxonomic abundance analysis)')
alphaDivOutput <- alphaDiv(genus, method = 'simpson')
#>
#> 2024-06-26 14:47:56.312006 Received df table with 741 samples and 226 taxa.
#>
#> 2024-06-26 14:47:56.327334 simpson alpha diversity computation complete.
#>
#> 2024-06-26 14:47:56.332597 Alpha diversity computation completed with parameters method= simpsonSpecies Evenness
Species evenness describes the distribution of abundances across the species in a sample. Species evenness is highest when all species in a sample have the same abundance and approaches zero as relative abundances vary.
## get an alpha diversity ComputeResult
genus <- getCollection(microbiomeData::HMP_MGX, 'Shotgun metagenomics Genus (Relative taxonomic abundance analysis)')
alphaDivOutput <- alphaDiv(genus, method = 'evenness')
#>
#> 2024-06-26 14:47:56.428865 Received df table with 741 samples and 226 taxa.
#>
#> 2024-06-26 14:47:56.44727 evenness alpha diversity computation complete.
#>
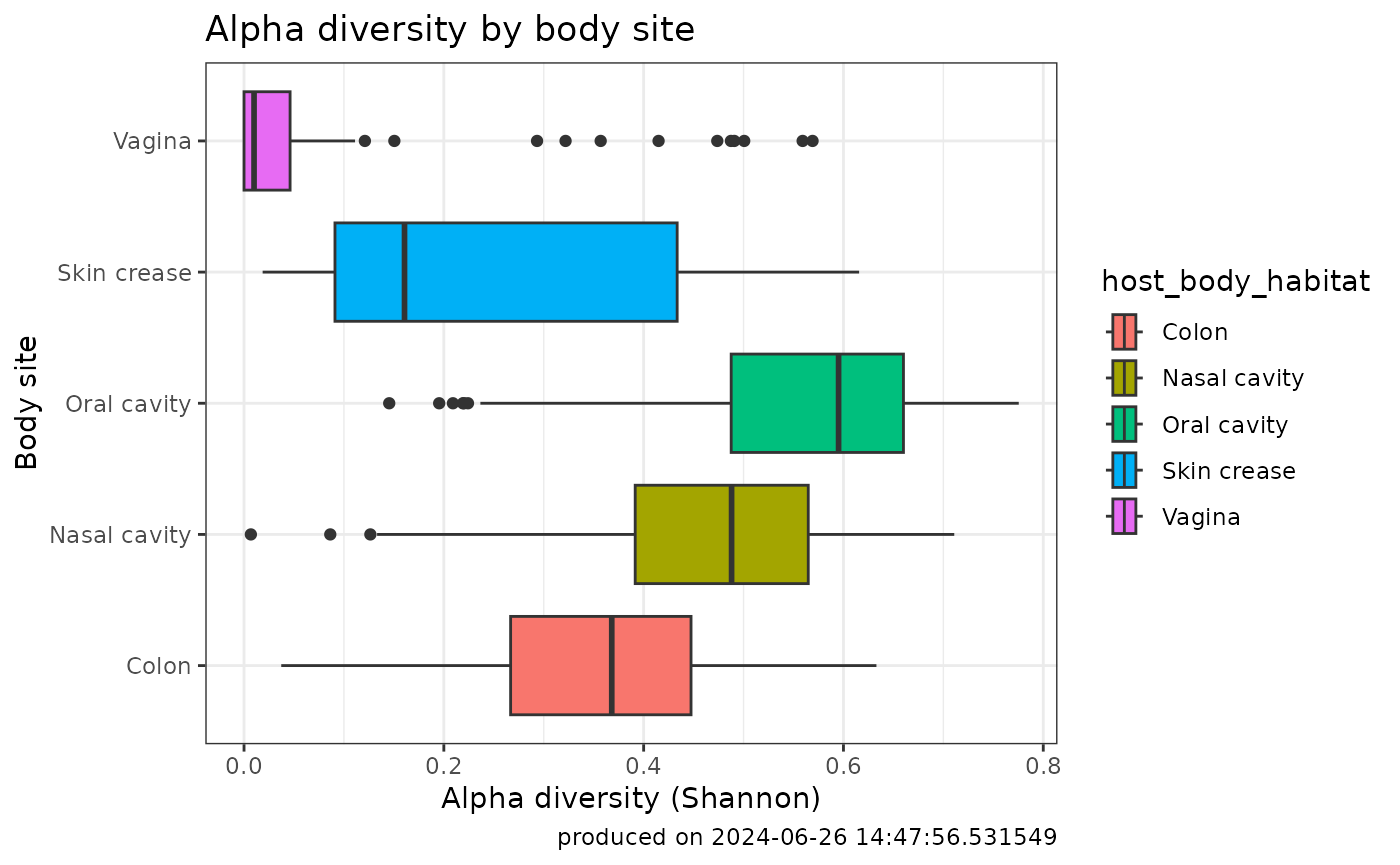
#> 2024-06-26 14:47:56.450666 Alpha diversity computation completed with parameters method= evennessVisualizing Alpha Diversity
Alpha Diversity is frequently visualized as scatter and box plots. Creating these types of plots can be done like the following:
## choose one or more metadata variables to integrate with the compute result
alphaDiv_withMetadata <- getComputeResultWithMetadata(
alphaDivOutput,
microbiomeData::HMP_MGX,
metadataVariables = c('host_body_habitat')
)
## plot the compute result with integrated metadata
ggplot(alphaDiv_withMetadata) +
aes(x=alphaDiversity, y=host_body_habitat, fill=host_body_habitat) +
geom_boxplot() +
labs(y= "Body site", x = "Alpha diversity (Shannon)",
title="Alpha diversity by body site",
caption=paste0("produced on ", Sys.time())) +
theme_bw()